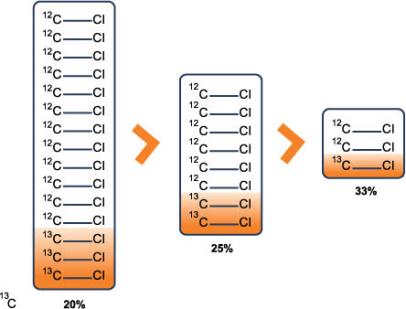
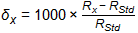3.3.3.1 Site Characterization
B) Are contaminant-degrading microorganisms active?
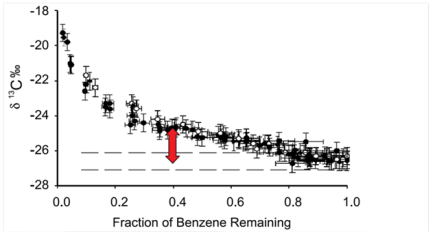
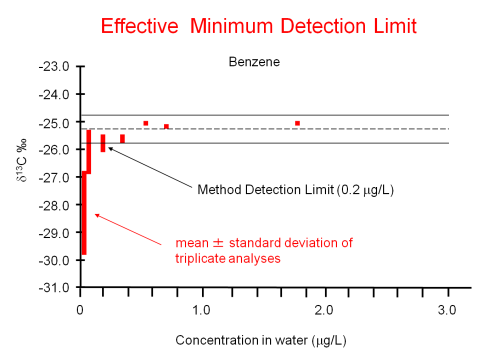
A release at a site contained benzene. After this initial release, contaminant concentrations were observed to decline over time. In addition, sulfate concentrations were lower than background in the core of the plume, indicating sulfate reducing conditions. Reports suggest that benzene degrading organisms are known to exist and be active under sulfate reducing conditions (Lovley et al. 1995). Moreover, the biodegradation of benzene by sulfate reducing strains has been reported to cause isotopic fractionation of carbon in laboratory samples (Mancini et al. 2003). Therefore, to determine if benzene was being degraded (as opposed to diluted), the δ13C values of benzene in groundwater were measured along a flow path downgradient of a suspected source at the site. The resulting δ13C data for each well are displayed in Figure 3-10.
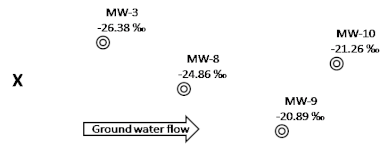
Figure 3-10. Map of an area in the site contaminated with benzene. The δ¹³C of the benzene was measured at each of the wells shown and the results are indicated on the map. Also note that the location of the source is marked with an “X”.
Source: Microseeps, Inc. Used with permission.
The combination of low sulfate concentrations, declining contaminant concentrations, and the increase in δ13C presented in Figure 3-10 suggests that sulfate-reducing benzene degrading microorganisms are active in this area.
D) Is biodegradation occurring?
Benzene Biodegradation
The example for Question B, showing the analysis of δ13C in benzene, answers the question of whether microorganisms are active and also provides clear evidence that biodegradation of benzene is occurring at the site. The increasing values of δ13C in benzene with distance from the source are evidence for biodegradation. With knowledge of groundwater flow rates and the relevant stable isotope enrichment factor (epsilon value) for carbon during benzene biodegradation by sulfate-reducing strains (Mancini et al. 2003), estimates of biodegradation rates can be obtained using stable isotope results. See USEPA guidance (USEPA 2008a) for a detailed description of the method and constraints for determining degradation rates using CSIA results.
TCE Biodegradation
At a site, the primary contaminant is TCE, and it is unclear from the concentration and geochemistry data whether TCE is biodegrading. The TCE was released into a fractured rock aquifer where distance from the source was not necessarily proportional to the time since release. CSIA was performed to see if there was evidence that biodegradation was occurring. An example subset of the results is presented in Table 3-3. In several wells (for example, MW4) the δ13C in TCE was higher, or more positive, than the highest currently published value of δ13C in TCE which is -24.5 ‰ (Aelion et al. 2010). In other locations, (for example MW2) the δ13C for TCE was significantly higher than similar values in the other wells (for example MW1), but not as high as in MW4. Historical records revealed all the TCE in this area was from the same source and the heavier (more positive δ13C) TCE was made more positive by degradation. The observation of TCE with δ13C heavier than the heaviest published literature value for product and of a range of δ13C values that are increasingly heavier than that found near the apparent source (well MW1 with highest residual concentrations) indicate that degradation has been active and that fractionation of δ13C in TCE has occurred or is currently occurring.
These data should be combined with analysis of geochemistry, relevant intermediate products, including cis-DCE, VC and ethene, and appropriate microbial analyses, such as qPCR for mccartyi (Dhc) to provide additional supporting evidence for TCE biodegradation. The data suggest that the degradation occurring is biodegradation. This is supported by qPCR and the appearance of transformation products. Additionally, the observation of sulfide (as would be expected for respiration through sulfate reduction) and/or the presence of methane concentrations above 1,000 μg/l (as would be expected for methanogenesis) further support the conclusion of biodegradation.
Table 3-3. CSIA (δ¹³C) results for selected example wells
|
MW1
|
<2
|
1400
|
849
|
-28.91
|
|
MW2
|
2.3
|
750
|
125
|
-25.95
|
|
MW3
|
2.1
|
3300
|
21
|
-23.82
|
|
MW4
|
3.8
|
180
|
6.6
|
-21.04
|
TCE Biodegradation
At another site with TCE contamination, despite decreasing concentrations of TCE, the CSIA values were not significantly different than the heaviest of the published values. There were three potential explanations for this result:
- Degradation of TCE was proceeding, but the biodegradation mechanism active in the subject location did not have a large enough enrichment factor to observably change the isotopic ratio. Literature values of enrichment factors (Aelion et al. 2010) can typically be used to see if this is possible. For example, the biodegradation mechanism could be sMMO catalyzed co-metabolism. Under this condition, it may be more appropriate to confirm the biodegradation with EAP or qPCR.
- Degradation was proceeding, but it was masked by the introduction of undegraded TCE into the dissolved phase. This typically occurs in the presence of NAPL, and there is NAPL at this site in some of the areas where the contaminants were not observed to be heavier than the heaviest published value. In a slowly recovering aquifer, back diffusion of contaminants can continue to be an issue (Sale et al. 2008). Because degraded contaminants are continually replaced by new ones diffusing from the geological matrix, the decline in concentration is a poor measure of attenuation. In an aquifer that is otherwise “clean” the concentration of the intermediate products may be too low to measure. In this situation, CSIA provides powerful insight into an aggressive biodegradation that traditional characterization measures of contaminant concentration may miss.
- No degradation was occurring, rather only minimally fractionating attenuation mechanisms such as dilution and diffusion were responsible for the observed decline in TCE concentrations.
Based on the data, it could not be concluded that biodegradation was occurring at this site. See Aelion et al. 2010; USEPA 2008a; Gray et al. 2002; Song et al. 2002; McLoughlin et al. 2013a; Palau et al. 2010; Morrill et al. 2009.
E) Is the contaminant attenuating abiotically?
At a site, leaded fuel had been released and the fuel additive 1,2-dibromoethane (EDB) was detected significantly above the regulatory MCL of 0.05 μg/L (USEPA 2008b). The EDB concentrations were declining over time, but the mechanism of decline was unclear based on the site data. Analysis of δ13C in EDB collected from several wells on-site was conducted using established methods to evaluate whether degradation could be confirmed based on stable isotopic ratios. The results from the analysis are presented in Table 3-4.
Table 3-4. EDB concentrations and δ¹³C of EDB for selected wells (USEPA 2008b)
|
MW5
|
15
|
-18.0
|
|
MW6
|
7.6
|
-10.5
|
|
MW7
|
2.7
|
-4.95
|
|
MW8
|
0.32
|
+11.0
|
Analysis of δ13C in EDB revealed significant isotope fractionation as a function of distance from the spill, so it was clear that the concentration decline was due at least in part to a degradative process, rather than just dilution or dispersion. The EDB was present in groundwater that was anoxic and has a low oxidation-reduction potential, presumably due to biodegradation of the fuel hydrocarbons. However, EDB degradation has been reported to occur in anoxic environments through both biological processes (Maymo-Gatell et al. 1997) and via abiotic transformation with hydrogen sulfide (H2S) (Schwartzenbach et al. 1985) and iron sulfide (FeS) (USEPA 2008b).
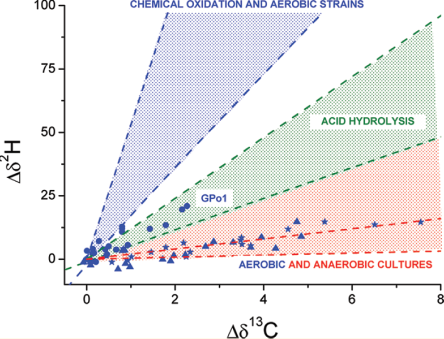
qPCR results showed no degradation capacity despite the application of a wide variety of probes, yet attenuation appeared to be occurring. CSIA results showed that the δ13C of the contaminants were heavier than the heaviest published values. Combined, this strongly suggests abiotic transformation. Further, the fractionation factors (ε values) for carbon during biological and abiotic degradation as shown in Figure 3-11 are such that the very enriched values observed from the field samples strongly suggest abiotic EDB degradation (USEPA 2008b). For additional information on isotope enrichment factors, see Section 3.3.4.1.
For more information on this specific question, see USEPA 2008b; VanStone 2004; Liang et al. 2007; Jeong et al. 2011; Hofstetter et al. 2007; Elsner et al. 2007; Poulson and Naraoaka, 2002).
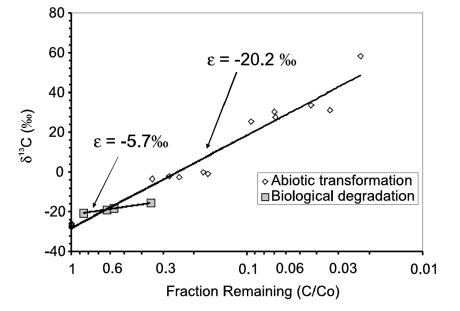
Figure 3-11. δ¹³C of EDB versus the fraction of EDB remaining for a biological study (Henderson et al. 2008) and another study in which the EDB was transformed abiotically.
Source: USEPA 2008b.
F) Are multiple sources contributing to the contamination?
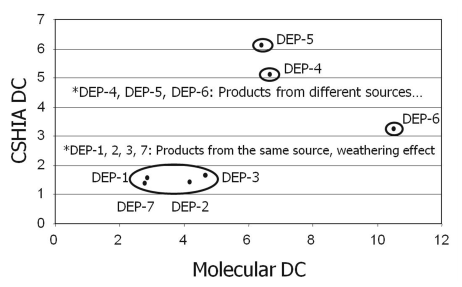
At a site perchlorate was detected in a number of monitoring wells at concentrations, ranging from a few ug/L to several mg/L. Some of the monitoring wells with high concentrations were clearly in an area of the site where propellants were discarded, and the source was easy to identify. However, several other wells with low concentrations of perchlorate were upgradient and sidegradient of this location, and did not have any other anthropogenic contaminants. Based on these data, stable isotope analysis of Cl and O in perchlorate was conducted in the primary plume location and for several of the upgradient and sidegradient wells to determine if multiple sources of perchlorate may be present at the site. The isotope data revealed that the δ18O, δ17O, and δ37Cl values of perchlorate from the primary plume were consistent with values typical for synthetic perchlorate, while the same isotopic ratios for the upgradient and sidegradient wells indicated a secondary low-level source, presumably derived from the past application of natural Chilean nitrate fertilizers (later determined to contain naturally occurring perchlorate) in the region during its past history as agricultural area. An example of the differing isotopic ratios for these sources is provided in Bohlke et al. 2009, and in Case Study A.1 on this topic.
For more information on this specific question, see Bohlke et al. 2005; 2009; Sturchio et al. 2006; 2012, Jackson et al. 2010.
G) If there is a potential for multiple sources, can the sources be distinguished?
Perchlorate
For perchlorate, this question is addressed in Question F. Synthetic and natural sources of this anion can be readily distinguished by stable isotope analysis of Cl and O, although it is much more difficult to discriminate synthetic sources from each other, as values of δ17O and δ37Cl differ very little among synthetic sources (Sturchio et al. 2006).
TCE
For chlorinated solvents, such as TCE, a number of potential situations exist in which multiple sources may be an issue. Most commonly, multiple sources are an issue when one or more sources are contributing to a groundwater plume, or when indoor air is impacted either by vapor intrusion from a plume under the property, or by commercial products brought into a home by the occupants. Both cases were observed at the example site.
Differing δ13C values have been used to discriminate sources of chlorinated ethenes (see Case Study A.3). For the purposes of this example, assume that similar analyses were conducted at one area of the site and showed multiple sources of TCE based on CSIA and supporting chemical concentration and hydrogeological information. Once multiple sources were identified, one of the important questions for wells between the sources was “how much of the contamination is from each source?” CSIA can be used to answer this question, but there are two very different applications of that question. One is for source apportionment in water, the second is for source apportionment in vapor (vapor could be ambient air or soil-gas).
Source Apportionment for a Water Sample
Source apportionment for TCE and other chlorinated solvents is most easily accomplished if biological or abiotic degradation of the parent compound has not occurred. In that case, it can be assumed that the observed δ13C (and δ37Cl values if available) for TCE in a well with mixed sources is just a concentration weighted average of the differing δ values of the individual sources. As an example, in one area of the site where two separate sources were identified, one off-site and one on-site, there was a downgradient well that was contaminated, but the contribution of each source to this contamination was unclear. CSIA was used in an attempt to trace the origin of the contamination and discern what percent of that impact was due to the on-site source and what percent of the impact was due to the off-site source. The site layout is presented in Figure 3-12 and the CSIA results are presented in Table 3-5.
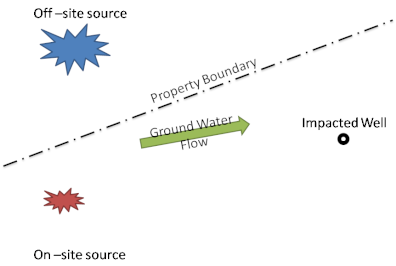
Figure 3-12. The area layout for an example site using CSIA to apportion source contributions.
Source: Microseeps, Inc. Used with permission.
In the impacted well, the δ13C was between the δ13C values of the two sources and the δ37Cl value also was between those of the two sources. In this case, and with no evidence of biological or abiotic degradation of TCE at the site, the contribution of source X is Fx and that of source Y is Fy and the linear relationship for the two sources is as follows:
Equation 3-2:
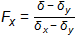
Equation 3-3:
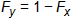
where δ is δ13C or δ37Cl for TCE from the well in question. Using the data in Table 3-5, the contribution of the off-site well is 80% for carbon. This value is corroborated by a similar calculation indicating the same contributions when chlorine is used.
Table 3-5. CSIA results for the example of apportionment in water
|
Off-site source
|
-30
|
-2
|
|
On site source
|
-25
|
+3
|
|
Impacted Well
|
-29
|
-1
|
Source Apportionment for a Vapor Sample
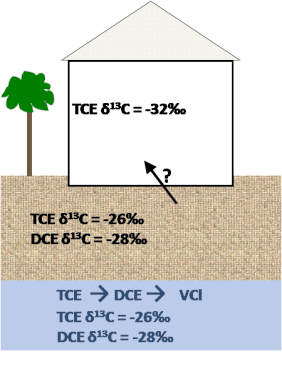
In a home situated above a plume on this site where TCE was being remediated by bio-augmentation and bio-stimulation, TCE was detected in the ambient air at concentrations above the action limit. However, the concentrations were sporadic and the plume shrinking, so it was believed that the source of the contaminant to the indoor air was not vapor intrusion. To better understand this situation, samples were collected from the air in the home and analyzed for δ13C and δ37Cl of the TCE. The TCE was found to be heavier than any published values and such fractionation could only come from degradation. Such degradation is clearly occurring in the treated groundwater plume based on isotope data and supporting parameters, but similar degradation is not expected for any airborne TCE brought into the home via consumer products. This result strongly implicates vapor intrusion from the groundwater plume as the cause of the indoor air concentrations. See Case Study A.2.
For more information on this specific question, see McHugh et al. 2011; Hunkeler et al. 2011; and Bouchard et al. 2008.